Solving Spatial Stochastic Systems in Julia
Ordinary Differential Equation (ODE) models in systems biology rely on the assumption that concentration is independent of location. When this assumption fails (which is fairly common as signaling molecules can exist below nano-molar concentrations), we need to either quantify the error that assumption creates or look for more sophisticated modeling approaches that can handle spatial dependences.
There are a growing number of popular methods that incorporate spatial dependence into dynamic systems (petri-nets, partial differential equations, agent based models, etc) but here I want to focus on spatial Stochastic Simulation Algorithms (SSAs). SSAs have been around for decades, going back to the famous Gillespie algorithm for chemical reaction systems. Instead of dealing with continuous values for concentration, the Gillespie algorithm assigns integer values to each species in the system. These counts are updated according to the chemical reactions connecting the individual species and the rate at which these reactions occur.
Extending this idea further, we can create some topological space (e.g. a 2D grid or a graph) and allow species to diffuse across that space. To demonstrate, let's try to extend the Lotka–Volterra equations (predator-prey model) to include a spatial dependence.
Discrete Lotka–Volterra System
For reference the Lotka–Volterra system is a set of two ODEs that describe the population dynamics between prey (like a rabbit) and predator (fox).
The variable DiffEqJump.jl and Cataylst.jl, packages that are a part of the SciML ecosystem.
using DiffEqJump, Catalyst, Plots
#"Reactions" for the Lotka–Volterra equations
#Note: β̄ = β - δ
LV_model = @reaction_network begin
α, x --> 2x
β̄, x + y --> y
δ, x + y --> 2y
γ, y --> ∅
end α β̄ δ γ
p = (2.0, 0.02, 0.02, 1.06) #(α β̄ δ γ)
u₀ = [100, 100] #100 prey and 100 predators
tspan = (0.0,20.0) #time span
prob = DiscreteProblem(LV_model, u₀, tspan, p)
jump_prob = JumpProblem(LV_model, prob, RSSA()) #rejection stochastic simulation algorithm
sol = solve(jump_prob, SSAStepper())
plot(sol, framestyle=:box, title="Discrete Lotka-Volterra Simulation")
Here we see the usual oscillations that characterize these equations. With few predators, the prey population increases, which in turn provides more food for predators. As predators hunt, the prey population is decreases, making less food for predators causing their population decrease and the cycle repeats. Note, however, there is noise in the simulation that causes fluctuations in the peaks of each populations. This may be a more realistic model especially when the populations are low (a fox might not be able to find the few remaining rabbits). Population levels are also discrete so you can't have less than one animal alive. This is an interesting distinction from the continuous version of this model. If the prey population goes to zero, the oscillations will stop and all the predators will also disappear.
2D Discrete Lotka–Volterra System
To extend this model and include location information for prey and predators, I followed this tutorial by Vasily Ilin which requires a few additional bits of information:
- A grid or network to allow the animals to roam
- Hopping rates, which tells the solver how easily an animal can move between locations
- A mass action jump object to code the reactions from the Lotka–Volterra System
Putting everything together gives:
using DiffEqJump, Catalyst
#Create a grid for animals to live on
dim = (64,64) #size of grid
numberOfNodes = prod(dim) #number of locations
grid = CartesianGrid(dim) #grid object
#"Reactions" for the Lotka–Volterra equations
#Note: β̄ = β - δ
LV_model = @reaction_network begin
α, x --> 2x
β̄, x + y --> y
δ, x + y --> 2y
γ, y --> ∅
end α β̄ δ γ
#Record the number of species in the model (2 in this case)
numberOfSpecies = numspecies(LV_model)
#Create an initial grid for each species
prey = zeros(Int,dim)
prey[1:dim[1]÷2,1:dim[2]÷2] .= 5 #all prey in bottom left corner
predators = reverse(prey) #all predators in top right corner
#Input requires each row in initial condition to be flattened
u₀ = zeros(Int, numberOfSpecies, numberOfNodes)
u₀[1,:] = prey[:]
u₀[2,:] = predators[:]
#Parameters and timespan for the model
p = (2.0, 0.02, 0.02, 1.06) #α β̄ δ γ
tspan = (0.0,20.0)
#How easy is it for species to move between locations?
hopConstants = ones(numberOfSpecies, numberOfNodes) #all set to one for now
#Create a mass action jump object
reactantStoich = [filter(x-> 0 ∉ x, 1:numberOfSpecies .=> col) for col in eachcol(substoichmat(LV_model))]
netStoich = [filter(x-> 0 ∉ x , 1:numberOfSpecies .=> col) for col in eachcol(netstoichmat(LV_model))]
massActionJumps = MassActionJump(reactantStoich, netStoich; param_idxs=1:numparams(LV_model))
#Generate the JumpProblem
prob = DiscreteProblem(u₀, tspan, p)
alg = DirectCRDirect() #could use NSM()
jumpProb = JumpProblem(prob, alg, massActionJumps, hopping_constants=hopConstants, spatial_system = grid, save_positions=(false, false))
#Solve the JumpProblem
sol = solve(jumpProb, SSAStepper(), saveat=0.1)The most confusing part is probably the generation of the mass action jump and is best explained through example. The reactant stoichiometry matrix records which species are reactants for every reaction. Here we have 2 species and 4 reactions, meaning substoichmat(LV_model) will give the following 2×4 matrix:
The second column (for example) corresponds to the reaction: x + y --> y. Both rows have a one because both x and y are reactants. This needs to be converted into a vector of Pairs where the first number corresponds to the species and the second number to the value in the matrix. For the matrix above we would get:
4-element Vector{Vector{Pair{Int64, Int64}}}:
[1 => 1] # x --> 2x
[1 => 1, 2 => 1] # x + y --> y
[1 => 1, 2 => 1] # x + y --> 2y
[2 => 1] # y --> ∅There may be a better way to generate this structure from the @reaction_network directly, but I could not find it. Once you have these structures for the reactants and the net stoichiometry, the rest is just passing variables to the solver, which has been specifically tailored to deal with these types of problems.
Any SSA solver could solve this problem, but solvers like NSM() optimize the solve by dividing the system into sub-volumes. If a reaction or diffusion event occurs we only have to worry about updating a subset of the simulation saving a lot of computation.
To visualize the solution, we can make a quick animation:
using Plots, Printf
#Plot an animation of the pedators and prey interacting
anim = @animate for (currState,t) in tuples(sol)
currTime = @sprintf "Time: %.2f" t
p1 = heatmap( reshape(currState[1,:],dim), alpha=1.0, c=:Blues_9, clims=(0,400), framestyle = :box, aspect_ratio=:equal, xlims=(1,dim[1]),ylims=(1,dim[1]), xlabel="Prey", title=currTime)
p2 = heatmap( reshape(currState[2,:],dim), alpha=1.0, c=:Oranges_9, clims=(0,400),framestyle = :box, aspect_ratio=:equal,xlims=(1,dim[1]),ylims=(1,dim[1]), xlabel="Predators")
plot(p1,p2, layout=(1,2))
end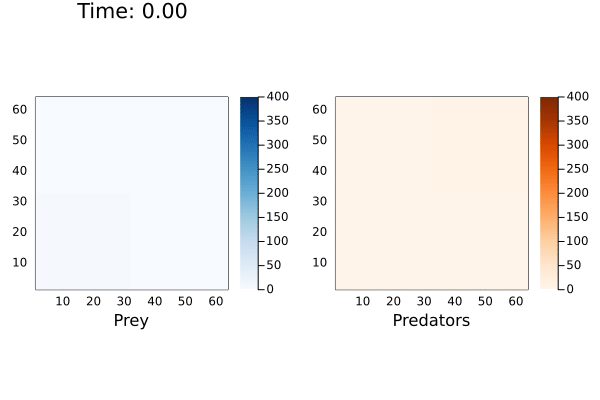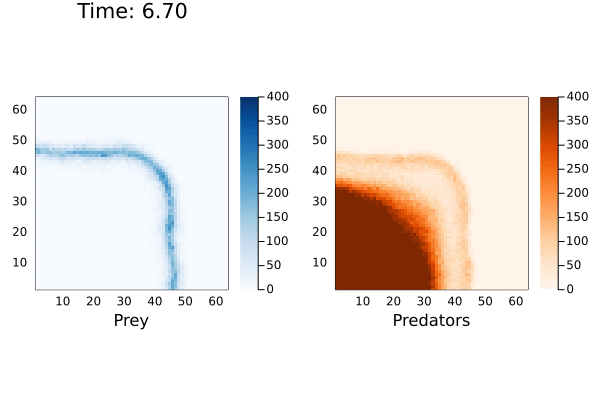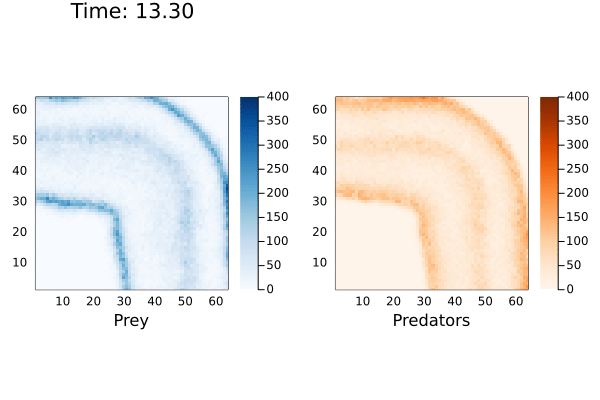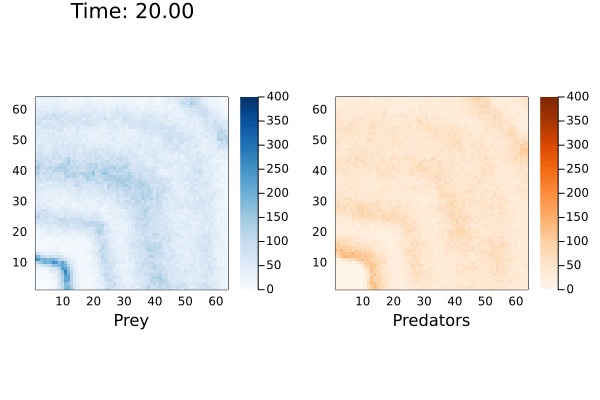
I've cut-off the maximum population size to 400 to better see the cyclic waves generated by the reaction. When playing with this model, I noticed the prey population exponentially increasing if there were no predators to consume them. This would cause the solver to hang indefinitely. Even in the simulation above the maximum number of prey reached over 18,000.
Some interesting questions I still have:
- How well does this scale with the number of species and number of equations?
- Could this be used to model cellular systems?
- A signaling molecule propagating across a cell or a virus relocating and assembling inside a cell?
- How would this model be extended to multi-scale systems (e.g. cell populations)?
- Are graphs/networks the best way to represent irregular geometry?
- Best way to handle boundary conditions?
- Could you use the hopping matrix to simulate membranes or other barriers to diffusion?
If you made this far, thanks for reading 😄!